添加日期:2017年3月21日 阅读:1775
3月17日,CFDA发布两大公告,一是总局公开征求《国家食品药品监督管理总局关于调整进口药品注册管理有关事项的决定(征求意见稿)》意见的通知;二是总局关于发布仿制药参比制剂目录(第*批)的通告(2017年第45号)。
这两大公告正与2017年1月《国务院办公厅关于进一步改革完善药品生产流通使用政策的若干意见》所提到的要优化药品审评审批程序,加快推进已上市仿制药质量和疗效一致性评价的政策内容相一致。
这也意味着,2015年8月起CFDA开始的一系列药品注册改革仍在继续,对应的目的在于加强药品注册管理,加快具有临床价值的新药和临床急需仿制药的研发上市,解决药品注册申请积压的矛盾。而且,改革不仅仅是国内化学药的注册改革,本次**将范围延伸到了进口药的注册审评。
仿制药参比试剂目录“两连发”
推荐参比制剂品种都有体现,有些品种增加规格,纳入较为**的产品注册地来源
3月17日,CFDA公布仿制药参比制剂目录(第*批)34个品种51个品规;紧接着3月20日即公布了仿制药参比制剂目录(第二批)21个品种33个品规,合计43个品种84个品规。
第*批CFDA公布的34个品种都是2018年年底前需要完成一致性评价的289目录内的产品。第二批公布的品种除了定点生产品种右佐匹克隆片,12个品种是需要完成一致性评价的289目录内的产品,其余都是289目录中的产品同通用名不同剂型的产品,如头孢呋辛酯干混悬剂和盐酸氨溴索缓释胶囊。
按CDE公布《2016年度药品审评报告》,目前仿制药一致性评价BE备案品种16个。咸达数据V3.2发现, BE备案已启动临床试验且登记号为2016年的产品有马来酸依那普利片、醋酸阿比特龙片、草酸艾司西酞普兰片、苯磺酸氨氯地平片、苯甲酸阿格列汀片、硫酸氢氯吡格雷片、甲苯磺酸索拉非尼片、琥珀酸索利那新片、泊沙康唑肠溶片、格列美脲片、替格瑞洛片、布洛芬缓释胶囊、富马酸替诺福韦二吡呋酯片、利奈唑胺片、利伐沙班片、他达拉非片,但这些产品并非都是一致性评价BE备案品种,其中苯磺酸氨氯地平片、硫酸氢氯吡格雷片、格列美脲片为289目录内产品,硫酸氢氯吡格雷片和格列美脲片在CFDA仿制药参比制剂目录中。
截至2017年3月20日,中国食品药品检定研究院分别在2016年8月、9月和11月分别推荐参比制剂品种,合计推荐17个品规(12个品种),以上品规数都在3月17日的《仿制药参比制剂目录(第*批)》51个品规中有所体现,其中头孢呋辛酯片在中检院的推荐名单中只有0.25g规格,CFDA第*批目录推荐增加0.125g规格,第二批目录中又增加了0.5g规格。
《仿制药参比制剂目录(第二批)》在中国食品药品检定研究院所推荐原有的品规中同通用名还增加规格的有辛伐他汀片增加了5mg和40mg,富马酸喹硫平片增加了0.2g、0.3g和50mg。盐酸特拉唑嗪片增加了1mg和5mg,马来酸依那普利片增加了25mg。硫酸氢氯吡格雷片增加300mg。
值得注意的是,CFDA发布的仿制药参比制剂目录都包括产品较为**的注册地来源。不知这是否意味着国内仿制药生产厂家需要根据来源所在国选择购买参比制剂?如若属实,则意味着表1的34个品种53品规可以选择国内进口产品做参比制剂。
表1 不同来源对应的产品名

数据来源:咸达数据V3.2
咸达数据V3.2发现, 1月《国务院办公厅关于进一步改革完善药品生产流通使用政策的若干意见》发布后,中检院在2月9日发布了美国FDA橙皮书(经过治疗等效性评价批准的药品)译文和日本橙皮书(医疗用医药品品质情报集)译文。这体现了CFDA及中检院为推动一致性评价而做的信息化方面推进的努力。
进口注册有望明显加快
对于此前利用进口药申报排队时间较长而抢首仿的旧3.1类企业打击较大
CFDA以往要求进口药物制造企业中国针对所有药品产品的进口流程均要求同时提交上市许可证明与进口注册申请,确认该药品获准在该国上市并销售之后,才能申请中国的临床试验授权。这将极大**新药进入中国上市的时间,也使得中国的患者无法使用全球市场*具创新性的药物。相比之下,在其他国家,原产国临床试验可与申请国临床试验同时进行。
表2 对比原药品注册管理办法
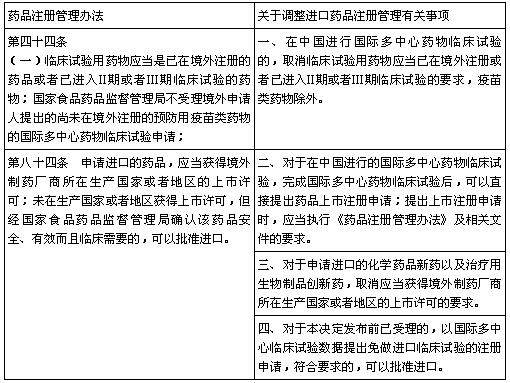
(数据来源:咸达数据V3.2)
按照旧化学药注册分类,以往进口药的注册分类不是1类就是3类,对应现化学药新分类的5.1类和5.2类。其中境外申请人在中国进行国际多中心药物临床试验IMCT(International Multicenter Clinical Trial)的还需要向国家食品药品监督管理局提出申请。
对于IMCT药物而言,多为新药,没有境外制药厂商所在生产国家或者地区的上市许可证书CPP(certificate of a pharmaceutical product),很难算旧3类药。于是,对于申报进口1类药物的生产厂家来说,正常的流程是需要先进行一次临床许可的申请,然后再完成一套完整的Ⅰ期、Ⅱ期、Ⅲ期临床试验,紧接着申请上市申请新药申请(NDA),*后得到进口药品注册证(IDL)。
鉴于走完整的Ⅰ~Ⅲ期所花的时间和费用成本都较高,而且*后还要等CPP结果才能申报生产。因此,进口申报往往旨在国外已获得CPP的前提下,在国内申报IMCT的部分临床方案,如Ⅱa、Ⅱb、Ⅲ期的PK、单独使用、联合用药、对比研究……总而言之,同样的品种申请多个不同目的的IMCT获得多个临床CTP登记号。病例数目很明显就是朝着满足中国对进口药品3类临床试验要求去的。在完成各种满足对进口药品3类的临床试验后,争取采用CDE的创新药物审评程序认定IMCT可以走特殊审批的流程报NDA免临床而直接获得IDL。
所谓“两报两批”,即按照“IMCT申请-临床试验-JXHL申请(免临床)-同意NDA免临床并获得IDL”这样的程序。按照这种程序操作下来,申请IMCT的临床CTP登记号预计需要10个月左右(进入特殊审批程序),临床试验3~5年,验证性临床试验申请(免临床)需要24~26个月,乐观估计,从申报IMCT到获得IDL,需要8~10年。
随着IMCT越报越多,新药临床试验申请(IND)的积压开始越来越多,CDE审批的时限也越来越长。CFDA/CDE于是要求进口药品注册必须严格按照《药品注册管理办法》规定的程序“三报三批”,即“IMCT申请-临床试验-JXHL进口新药化学药临床试验申请(免临床)-进口新药化学药生产JXHS申请-获得进口药品注册证IDL”这样的程序。按照CFDA/CDE的审评速度,“三报三批”意味着增加了2.5年甚至更久的审批时间,使得IMCT途径取得的IDL推后2年甚至更久。
CFDA本次改革流程主要是改变了新化学药注册分类5.1类的注册规则,IMCT的Ⅰ期临床将有可能也会纳入中国的临床案例数。更重要的是,目前在排队申报生产的以往积压的且免临床的进口药有望在近期快速上市,前提是临床自查核查要过关。
此新规对于国内企业此前由于进口药的申报排队时间较慢从而抢首仿旧3.1类药品的企业打击较大。首先,原研药将会在近期全部上市意味着抢在原研药前上市基本不可能;其次是原研药的专利期保护下,2015年至2016年批量获批临床的国内仿制药企业即使在2016年至2017年启动临床,可能也要等专利期前1年申报生产。况且,按照新规,仿制药还要必须过一致性评价试验,原来申报旧3.1类的企业还不如直接走BE备案重新排队更快更省钱。
总结>>>
无论是一致性评价信息公开,还是进口药品注册管理有关事项的调整,都表达CFDA对药品注册审评积压改革的积极态度。3月15日药审中心发布加快推进eCTD项目建设的公告,意味着2017年底起所有化学仿制药的申报和审评都在eCTD系统上实施是非常有可能的。CFDA和CDE的目标是往国际化生产,难免下一步的改革就是全面学习FDA采取创新药物及其制剂的申请(NDA)、仿制药的申请(ANDA)和OTC(非处方药)的申请模式。
此外,化学药改革完成后,中药和生物制品的改革也会随之而来。目前只是阶段性改革,国内企业立项切勿时刻紧盯国家药品注册政策导向并以之为主要理由,而应放长目光,早日往国际化和合规化方向发展方为正道。
文章来源:
1.凡本网注明“来源:1168医药招商网”的所有作品,均为广州金孚互联网科技有限公司-1168医药招商网合法拥有版权或有权使用的作品,未经本网授权不得转载、摘编或利用其它方式使用上述作品。已经本网授权使用作品的,应在授权范围内使用,并注明“来源:1168医药招商网http://www.1168.tv”。违反上述声明者,本网将追究其相关法律责任。
2.本网转载并注明自其它来源(非1168医药招商网)的作品,目的在于传递更多信息,并不代表本网赞同其观点或和对其真实性负责,不承担此类作品侵权行为的直接责任及连带责任。
3.其他媒体、网站或个人从本网转载时,必须保留本网注明的作品第一来源,并自负版权等法律责任。
4.如涉及作品内容、版权等问题,请在作品发表之日起一周内与本网联系,否则视为放弃相关权利。联系邮箱:1753418380@qq.com。

【适用范围】用于缓解颈、肩、腰、腿及闭合性软组织疼痛、肿胀等不适症状人群的物理冷敷。【使用方法】外用。将本品适量直接涂抹于不适部位,轻轻按摩2-3分钟,每日2-3次。

【适用范围】用于缓解颈、肩、腰、腿及闭合性软组织疼痛、肿胀等不适症状人群的物理冷敷。【使用方法】外用。将本品适量直接涂抹于不适部位,轻轻按摩2-3分钟,每日2-3次。
 粤公网安备 44011102000390号
粤公网安备 44011102000390号